- Article
- Source : Campus Sanofi
- 1 févr. 2023
L’insuffisance rénale chronique et terminale dans la maladie de Fabry
La maladie de Fabry, liée au chromosome x, est une pathologie causée par un déficit enzymatique en α-galactosidase A. Ce déficit entraine une accumulation de glycosphingolipides (Gb3) dans les lysosomes engendrant des atteintes tissulaires et organiques.1
Découvrez comment les accumulations de Gb3 dans les cellules rénales peuvent aboutir à une insuffisance rénale chronique. Votre rôle en tant que professionnel de la santé est essentiel pour éviter la survenue d’une insuffisance rénale terminale.
L’atteinte rénale dans la maladie de Fabry
Les dépôts progressifs de Gb3 dans les différents types de cellules rénales provoquent une ischémie rénale progressive et favorisent l’inflammation et la fibrogénèse.1,2
Au niveau rénal, les accumulations de Gb3 se localisent3
- Dans toutes les cellules glomérulaires
- Aux capillaires péritubulaires
- Aux capillaires endothéliaux
- Dans les cellules musculaires lisses
- Dans les cellules tubulaires distales
Les atteintes rénales initiales donnent lieu à une insuffisance rénale chronique (IRC) chez :
• 84 % des hommes4
• 35 % des femmes5
L’insuffisance rénale chronique peut progresser vers une insuffisance rénale terminale (IRT).1

Devant une néphropathie d’origine inconnue, avez-vous pensé à une maladie de Fabry ?
La maladie de Fabry serait à l’origine de 0,3 à 1 % des insuffisances rénales dont la cause est indéterminée.7,8
Répartition des néphropathies initiales des patients dialysés en 2019
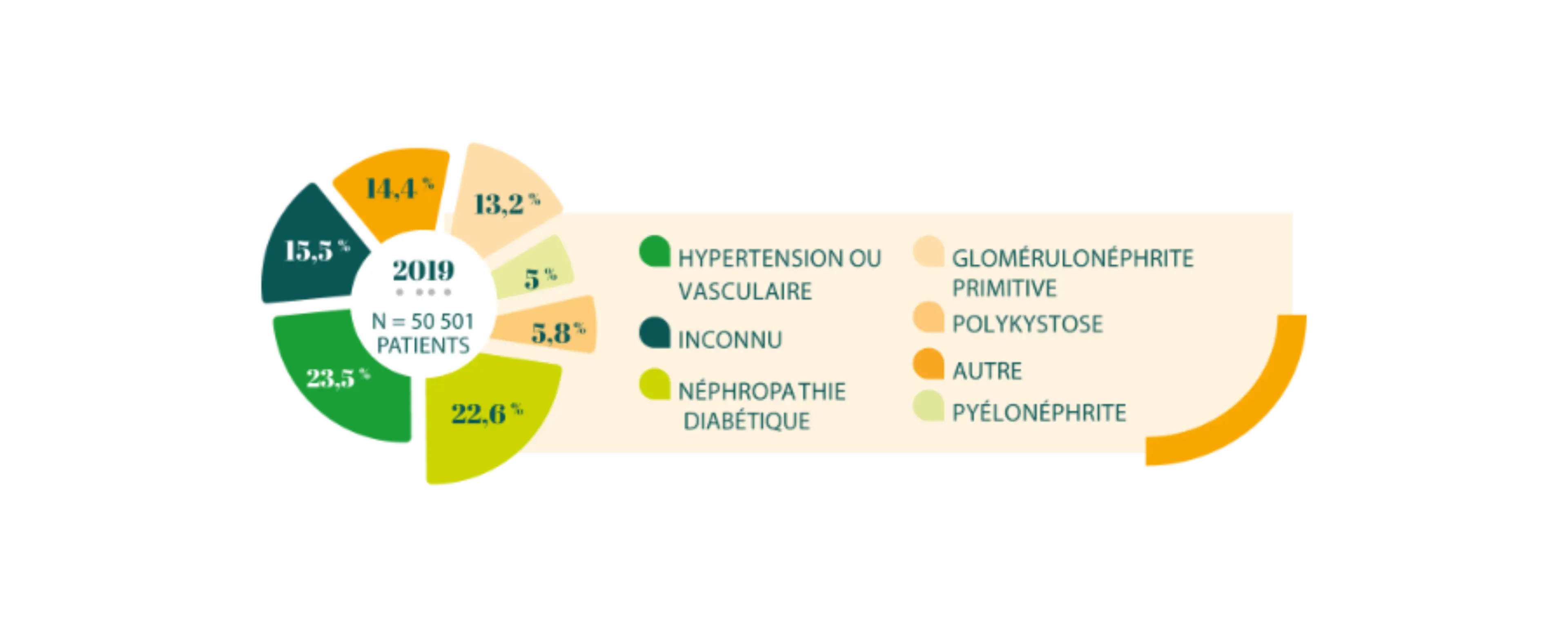
D’après : REIN - Rapport annuel 2019.
Pour 15,5 % des patients en dialyse, la cause de leur néphropathie est inconnue.9
Les néphropathies hypertensives / vasculaires ou diabétiques qui représentent la moitié des cas, n’excluent pas pour autant une maladie de Fabry.1,10
Comment exclure une maladie de Fabry avant la survenue de l’IRT ?
Lorsqu’une biopsie est pratiquée, elle peut faire évoquer une maladie de Fabry.1,11
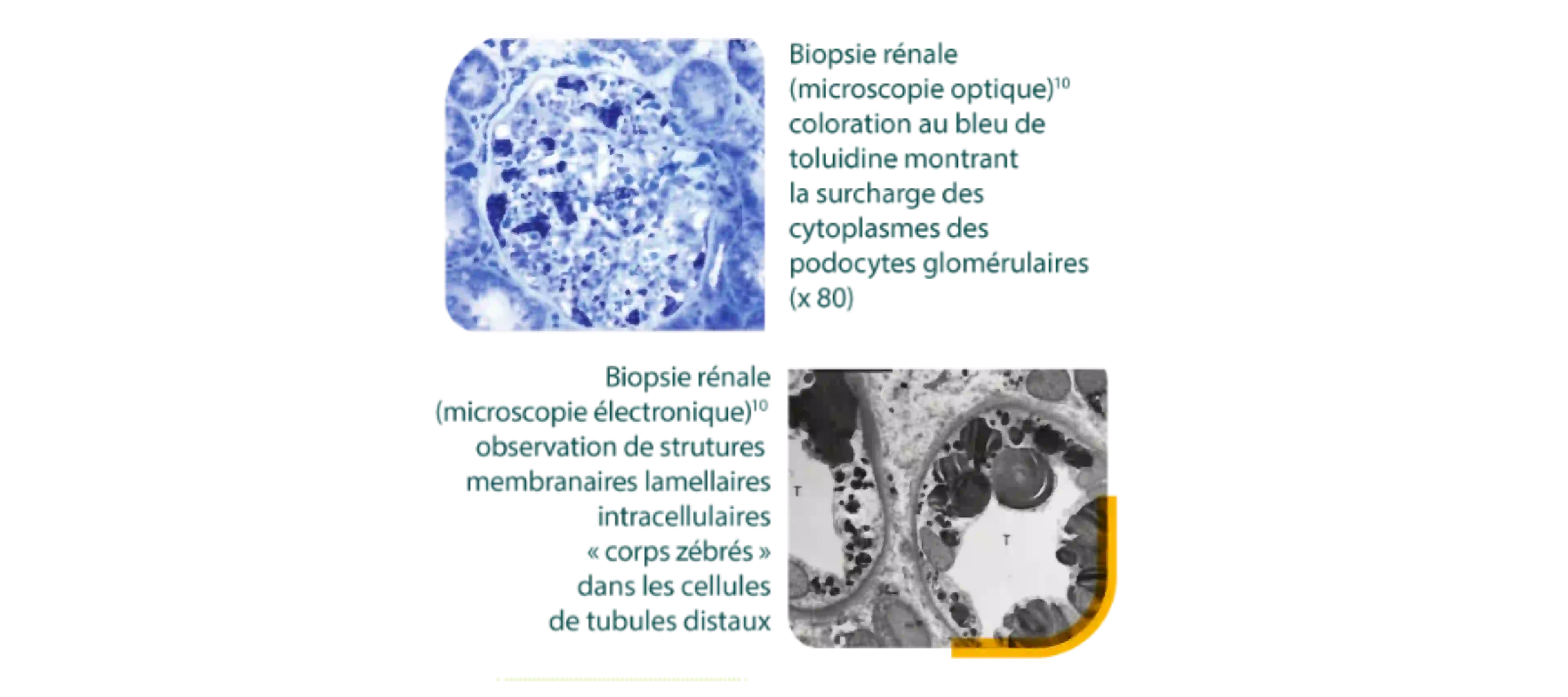
Images : Germain DP. Orphanet J Rare Dis 2010; 5: 30
Caractéristiques histologiques de l’atteinte rénale dans la maladie de Fabry.11
- Dépôts progressifs de Gb3 dans les différents types de cellules provoquant une ischémie rénale progressive.
- Vacuolisation cytoplasmique dans tous les types cellulaires (glomérulaires, cellules épithéliales tubulaires, myocytes, et cellules endothéliales).
- Corps myéliniques et corps zébrés (visible au microscope électronique), observés dans tous les types cellulaires rénaux.
Recommandations de l’ERA (European Renal Association) pour le diagnostic de la maladie de Fabry13

L’hypertension ne doit pas être considérée comme un critère d’exclusion puisque 50 % des patients atteints de la maladie de Fabry présentent une hypertension légère à modérée (principalement lorsque le DFGe < 60 mL / min / 1,73 m2).
Un diagnostic possible grâce à une simple prise de sang1
.png)
Pour les hommes
Mesure de l’activité α-galactosidase A dans les leucocytes ET/OU sur goutte de sang séché et la mise en évidence d’un variant pathogène du gène GLA.
.png)
Pour les femmes
Si mesure de l’activité α-galactosidase A déficitaire ET/OU lyso-Gb3 plasmatique élevé ET/OU forte suspicion clinique : réalisation d’un génotypage du gène GLA.
À RETENIR
- 84 % des hommes et 35 % des femmes atteints de la maladie de Fabry présentent une atteinte rénale4
- Une insuffisance rénale terminale pouvant se déclarer à partir de 40 ans8
- Présence concomitante possible d’une maladie de Fabry chez des patients hypertendus et/ou diabétiques15,16
- Diagnostic simple1
- Des traitements existent afin de prévenir la survenue de lésions irréversibles1
Références
- Protocole National de Diagnostic et de Soins Maladie de Fabry. HAS Novembre 2021.
- Najafian B, et al. JASN. 2020; 31:865-875.
- Wanner C, et al. Clin J Am Soc Nephrol. 2010 Dec;5(12):2220-8.
- MacDermot K.D, et al. J Med Genet. 2001; 38:750-760
- MacDermot K.D, et al. J Med Genet. 2001; 38: 769-775
- Waldek S, et al. Genet Med. 2009; 11(11): 790-796
- Van der Tol L, et al. J Med Genet. 2014 ; 51 : 1-9
- Bekri S, et al. Nephron Clin Pract. 2005 ; 101 :c33-c38
- REIN - Rapport annuel 2019. Agence de la biomédecine.
- Germain DP, et al. Mol Genet Genomic Med. 2018;1-12.
- Thurberg B.L, et al. Kidney International. 2002;62:1933 -1946.
- Germain DP. Orphanet J Rare Dis 2010; 5: 30.
- Terryn W, et al. Nephrol Dial Transplant. 2013;28(3):505-17.
- Eng CM. et al. J Invertit Metab Dis (2007);30:184–192.
- Sodré LSS, et al. Kidney Blood Press Res. 2017 ; 42 :1258-1265
- Nakao S, et al. N. Engl. J. Med. 1995 ; 333(5): 288-293
7000040865 - 02/2023