- Article
- Source : Campus Sanofi
- 1 févr. 2023
Qu'est-ce que la maladie de Fabry ?

La maladie de Fabry est une maladie génétique liée à une mutation du gène GLA sur le chromosome X. Cette mutation est à l’origine d’un déficit enzymatique en alpha-galactosidase A provoquant une accumulation de glycosphingolipides au niveau intracellulaire (Gb3) et plasmatique (lyso-Gb3).
Cette condition, débutant dès le plus jeune âge, engendre des atteintes multisystémiques et irréversibles, souvent associées à une morbi-mortalité élevée à l’âge adulte en l’absence de traitement.1
Signes cliniques de la maladie de Fabry
.png)
Parmi les atteintes cliniques de la maladie de Fabry, on retrouve1 :
- L’atteinte oculaire
Les patients peuvent présenter une cornée verticillée sans diminution de l’acuité visuelle ainsi que des opacités cornéennes. - L’atteinte auditive
Les patients peuvent présenter des signes d’hypoacousie et de surdité. - L’atteinte du système nerveux central
Les patients peuvent présenter un AIT (Accident Ischémique Transitoire), ainsi qu’un AVC (Accident Vasculaire Cérébral) précoces et récidivants. - La symptomatologie associée à l’atteinte cardiaque est l’hypertrophie ventriculaire gauche (HVG), les troubles de la conduction et du rythme ainsi que l’atteinte valvulaire (ex : insuffisance mitrale).
- Le patient atteint de la maladie de Fabry peut également présenter des troubles digestifs parmi lesquels les douleurs abdominales, les diarrhées et vomissements.
- Les patients atteints de la maladie de Fabry peuvent également présenter une atteinte cutanée sous la forme d’angiokératomes (papules violacées au niveau de l’aine).
- L’atteinte rénale est également une symptomatologie pouvant être rencontrée chez les patients atteints de la maladie de Fabry. Celle-ci peut se manifester par une microalbuminurie/protéinurie, insuffisance rénale chronique puis terminale conduisant à la dialyse et transplantation.
- Le système nerveux périphérique peut également être atteint avec présence de douleurs chroniques neuropathiques et acroparesthésies.
Des fièvres récidivantes, un dysfonctionnement sudoral (hypohidrose ou anhidrose), une intolérance à la chaleur et ou à l’exercice physique peuvent être retrouvés chez ces patients.
Enfin, des antécédents familiaux tels que l’atteinte rénale et/ou cardiaque et/ou cérébrovasculaire ainsi qu’un décès précoce et/ou inexpliqué peuvent alerter sur une maladie de Fabry.
La transmission de la maladie est liée au chromosome X touchant les hommes et les femmes.
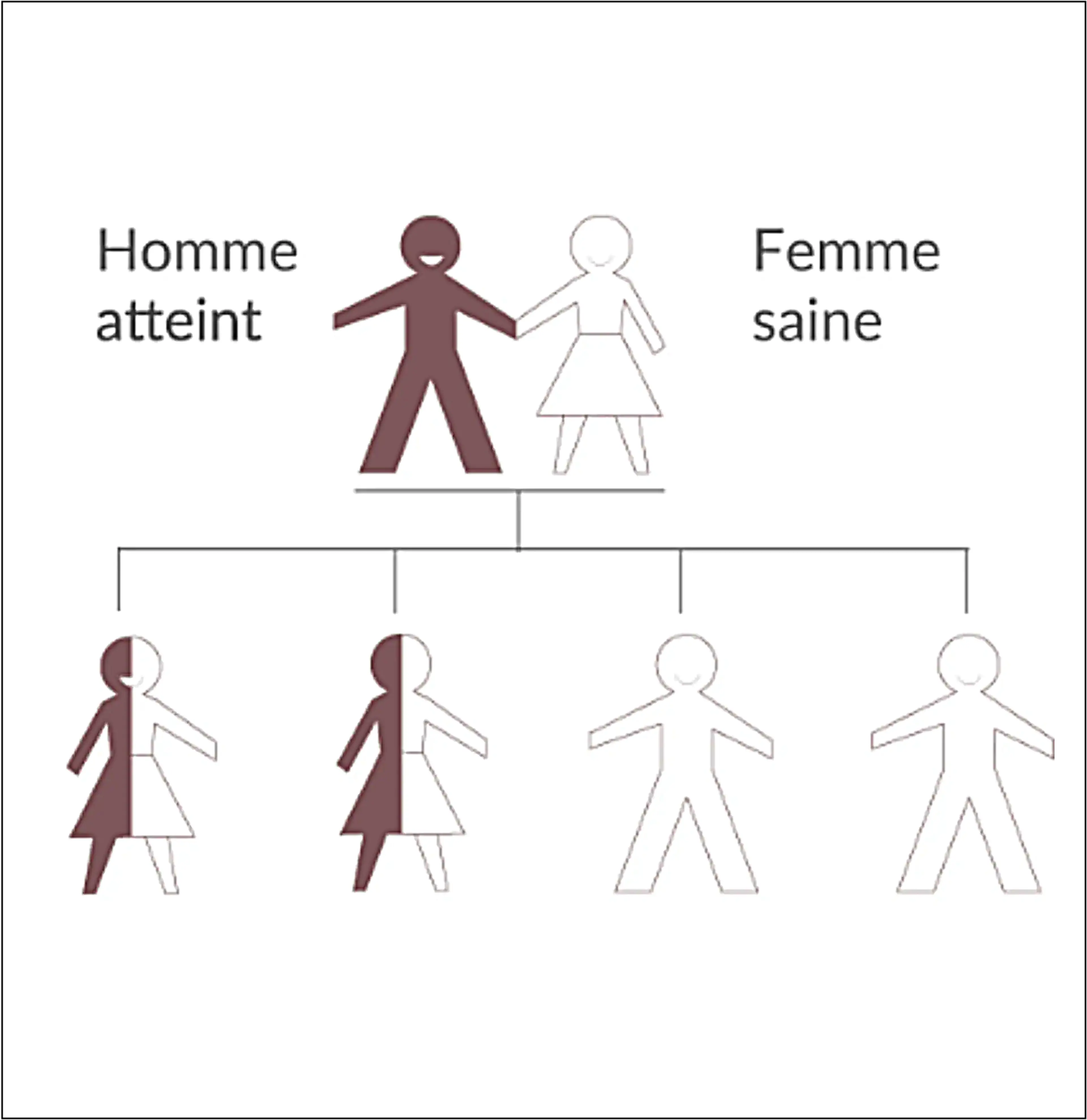
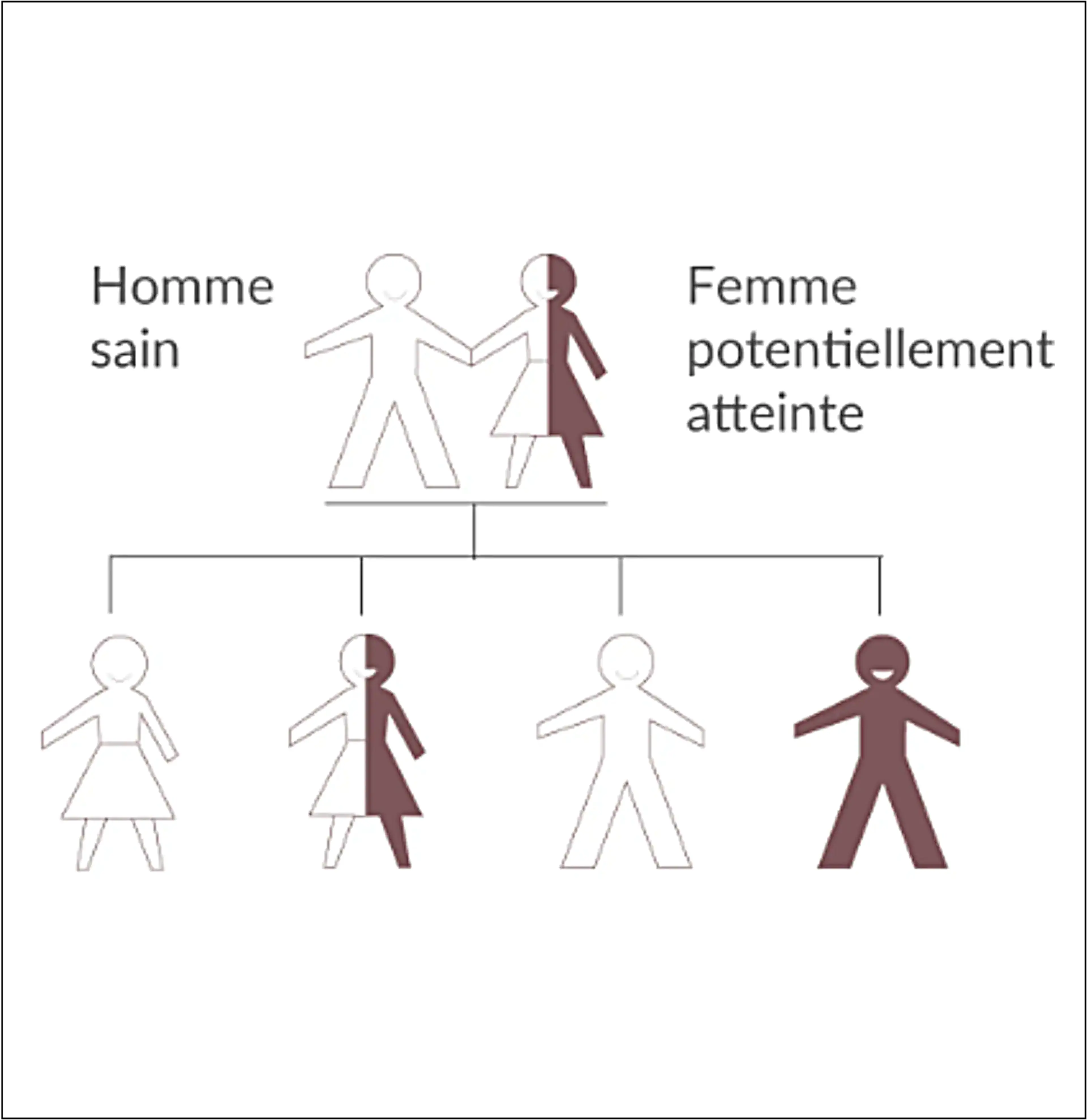
Un retard au diagnostic peut avoir des conséquences graves sur le devenir des patients atteints de la maladie de Fabry
- Retard au diagnostic médian* de2:
- 14 ans pour les hommes
- 19 ans pour les femmes
L’espérance de vie se retrouve donc diminuée de 15 ans chez la femme et 20 ans chez l’homme, par rapport à la population générale.3,4
*Par rapport aux premiers symptômes
Ne passez pas à côté du diagnostic simple, à l’aide d’une simple prise de sang
.png)
Pour l’homme
Mesure de l’activité α-galactosidase A dans les leucocytes ET/OU sur goutte de sang séché et la mise en évidence d’un variant pathogène du gène GLA.
.png)
Pour la femme
Si mesure de l’activité α-galactosidase A déficitaire ET/OU lyso-Gb3 plasmatique élevé ET/OU forte suspicion clinique : réalisation d’un génotypage du gène GLA.
Il existe des traitements spécifiques permettant d’éviter la survenue de lésions irréversibles pour les patients présentant un diagnostic confirmé de la maladie de Fabry. D’où l’importance d’un diagnostic précoce.¹
Références
- Protocole National de Diagnostic et de Soins Maladie de Fabry. HAS. Novembre 2021.
- Eng CM, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007 Apr;30(2):184-92.
- MacDermot KD, et al. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001 Nov;38(11):769-75.
- MacDermot KD, et al. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001 Nov;38(11):750-60.
7000040864 - 02/2023