- Article
- Source : Campus Sanofi
- 31 janv. 2023
Qu'est ce que la mucopolysaccharidose de type 1 ?

La mucopolysaccharidose de type 1 (ou MPS1) est une maladie génétique rare et sous-diagnostiquée. Elle existe sous 3 formes : Hurler, Hurler Scheie et Scheie1. Découvrez dans cet article les signes d'appels de la MPS1 et la conduite à tenir en cas de suspicion.
Les signes d’appel sont multisystémiques et hétérogènes. L’âge et l’ordre d’apparition des symptômes, leur sévérité et l’évolution de la maladie, sont propres à chaque patient1,2,3.
La mutation du gène IDUA induit un déficit enzymatique en α-L-iduronidase entrainant une accumulation de glycosaminoglycanes (héparane sulfate et dermatane sulfate)2,4,5.
Une prise en charge thérapeutique est possible : la greffe* et/ou le traitement enzymatique substitutif2.
| Un diagnostic précoce est essentiel pour optimiser la prise en charge et le devenir clinique des patients1. |
Les signes d’appel de la forme sévère (Hurler)
L’enfant est normal à la naissance et les premiers signes apparaissent dès les premiers mois de vie.1
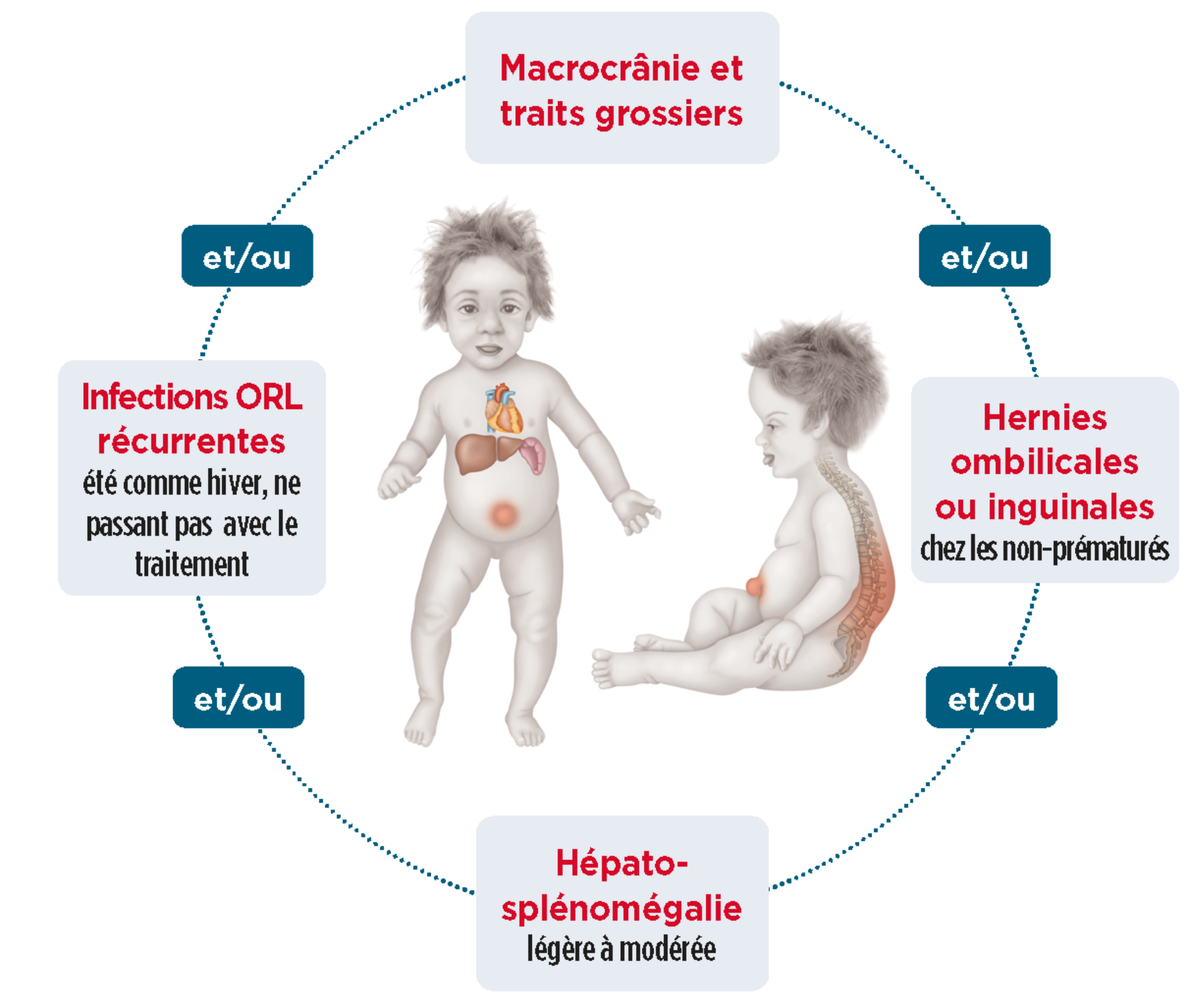
Descriptif complémentaire des principaux signes d'appelInfections ORL à répétition4
Hernies ombilicale et/ou inguinale4
Hépatosplénomégalie légère à modérée4
Dysmorphie / traits grossiers4
Liste non exhaustive. Autres signes fréquents : macrocranie, infections ORL à répétition, troubles du sommeil, dysostose multiple,...2,4 |
Les signes d’appel des formes atténuées (Hurler Scheie et Scheie)
L’apparition des premiers signes survient à partir de 3-4 ans.1
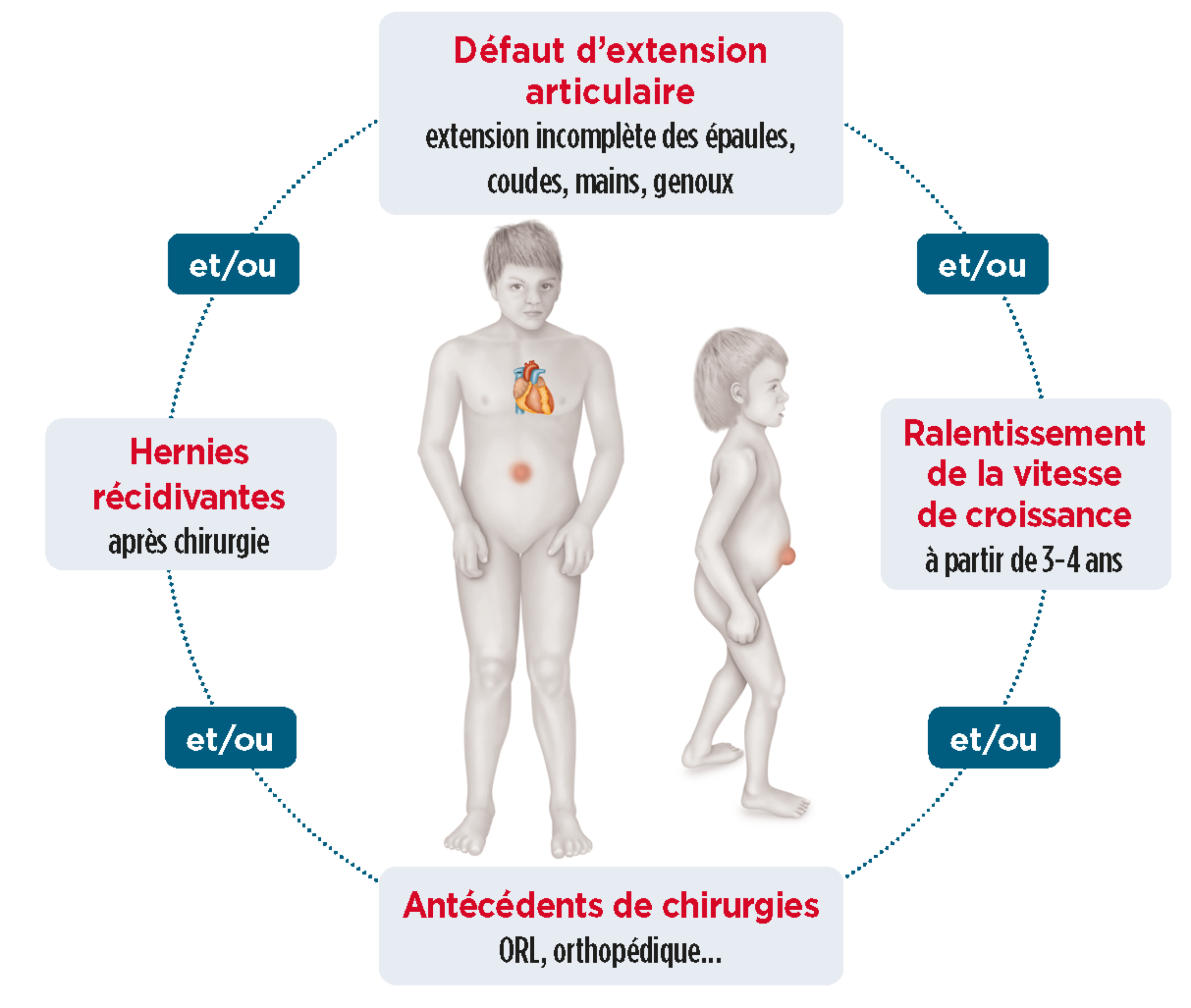
Descriptif complémentaire des principaux signes d'appelAntécédents de chirurgies ORL7
Limitation de l’amplitude articulaire8,9,10
Hernies ombilicale et/ou inguinale4
Ralentissement de la vitesse de croissance6
Liste non exhaustive. Autres signes fréquents : opacité cornéenne, hépatosplénomégalie, syndrome du canal carpien bilatéral, troubles du sommeil, valvulopathie, dysostose multiple, ...2,4 |
Conduite à tenir en cas de suspicion
Le diagnostic de certitude de la MPS1, simple et rapide, repose sur la mise en évidence d’un déficit profond enzymatique en α-L-iduronidase1,2.
Examens urinaires
|
Analyse quantitative : élévation des GAGs** Dans les formes atténuées, les taux urinaires de GAGs** peuvent être normaux ou peu augmentés |
Examen sanguin
|
Dosage de l’activité enzymatique résiduelle de l’enzyme α-L-iduronidase1,2 (dans les leucocytes, les fibroblastes, les amniocytes en cultures et les villosités choriales) La valeur de l’activité enzymatique ne permet pas de distinguer une forme sévère des formes atténuées |
La recherche de mutation du gène IDUA codant pour l’α-L-iduronidase (4p16.3) peut permettre, pour certaines mutations, de prédire la sévérité du phénotype2.
Plusieurs laboratoires spécialisés sont en mesure de réaliser le dosage urinaire des GAGs** et le dosage enzymatique permettant de confirmer le diagnostic de la MPS1.
* transplantation de cellules souches hématopoïétiques
** glycosaminoglycanes
Références
- Beck et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genetics in Medecine. 2014; 16, 759–65
- Protocole National de Diagnostic et de Soins MPS – HAS – Juillet 2016
- Moore D. et al. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008; 16, 3-24
- Guffon et al. La mucopolysaccharidose type 1 (MPS I): présentation clinique et traitement actuel. Médecine thérapeutique / Pédiatrie. 2003; 6 (2), 91-7
- Michaud M, Belmatoug N, Catros F, et al. Mucopolysaccharidoses :quand y penser ? [Mucopolysaccharidosis: A review]. Rev Med Interne. 2020; 41, 180-88
- Guffon et al. Growth impairment and limited range of joint motion in children should raise suspicion of an attenuated form of mucopolysaccharidosis: expert opinion. European Journal of Pediatrics. 2019;178, 593-603
- Arn et al. Characterization of Surgical Procedures in Patients with Mucopolysaccharidosis Type I: Findings from the MPS I Registry. J Pediatr. 2009 ; 154(6): 859-64
- Journeau et al. Raideurs articulaires chez l’enfant : est-ce toujours pathologique ? Pédiatrie pratique 291. 2017
- Rigoldi M, Verrecchia E, Manna R, Mascia MT. Clinical hints to diagnosis of attenuated forms of Mucopolysaccharidoses. Ital J Pediatr. 2018;44(Suppl 2):132.
- Tylki-Szymańska A, De Meirleir L, Di Rocco M, Fathalla WM, Guffon N, Lampe C, Lund AM, Parini R, Wijburg FA, Zeman J, Scarpa M. Easy-to-use algorithm would provide faster diagnoses for mucopolysaccharidosis type I and enable patients to receive earlier treatment. Acta Paediatr. 2018 Aug;107(8):1402- 1408. doi: 10.1111/apa.14417. PMID: 29797470; PMCID: PMC6055821.
7000040910 - 01/2023